



DNA-Sequenzklassifizierung basierend auf Milvus
Autor: Mengjia Gu, Data Engineer bei Zilliz, hat an der McGill University einen Master-Abschluss in Information Studies erworben. Zu ihren Interessen gehören KI-Anwendungen und Ähnlichkeitssuche mit Vektordatenbanken. Als Community-Mitglied des Open-Source-Projekts Milvus hat sie verschiedene Lösungen bereitgestellt und verbessert, wie etwa ein Empfehlungssystem und ein DNA-Sequenzklassifizierungsmodell. Sie liebt Herausforderungen und gibt niemals auf!
Einführung
Die DNA-Sequenz ist ein populäres Konzept sowohl in der akademischen Forschung als auch in praktischen Anwendungen, wie etwa Gen-Rückverfolgbarkeit, Artenidentifikation und Krankheitsdiagnose. Während alle Branchen nach einer intelligenteren und effizienteren Forschungsmethode verlangen, hat künstliche Intelligenz insbesondere im biologischen und medizinischen Bereich viel Aufmerksamkeit erregt. Immer mehr Wissenschaftler und Forscher tragen zu Machine Learning und Deep Learning in der Bioinformatik bei. Um experimentelle Ergebnisse überzeugender zu machen, ist eine gängige Option die Erhöhung der Stichprobengröße. Die Zusammenarbeit mit Big Data in der Genomik eröffnet ebenfalls mehr Möglichkeiten für Anwendungsfälle in der Realität. Die traditionelle Sequenzalignierung hat jedoch Einschränkungen, die sie für große Datenmengen ungeeignet machen. Um in der Realität weniger Kompromisse eingehen zu müssen, ist Vektorisierung eine gute Wahl für einen großen Datensatz von DNA-Sequenzen.
Die Open-Source-Vektordatenbank Milvus ist für massive Datenmengen geeignet. Sie kann Vektoren von Nukleinsäuresequenzen speichern und eine hocheffiziente Suche durchführen. Sie kann auch dazu beitragen, die Kosten von Produktion oder Forschung zu senken. Das auf Milvus basierende DNA-Sequenzklassifizierungssystem benötigt nur Millisekunden für die Genklassifizierung. Darüber hinaus zeigt es eine höhere Genauigkeit als andere gängige Klassifikatoren im Machine Learning.
Datenverarbeitung
Ein Gen, das genetische Informationen kodiert, besteht aus einem kleinen Abschnitt von DNA-Sequenzen, der aus 4 Nukleotidbasen [A, C, G, T] besteht. Es gibt etwa 30.000 Gene im menschlichen Genom, fast 3 Milliarden DNA-Basenpaare, und jedes Basenpaar hat 2 entsprechende Basen. Zur Unterstützung vielfältiger Anwendungen können DNA-Sequenzen in verschiedene Kategorien eingeteilt werden. Um die Kosten zu senken und die Nutzung von Daten langer DNA-Sequenzen zu erleichtern, wird k-mer in die Datenvorverarbeitung eingeführt. Gleichzeitig macht es DNA-Sequenzdaten einfacherem Text ähnlicher. Darüber hinaus können vektorisierte Daten Berechnungen in der Datenanalyse oder im Machine Learning beschleunigen.
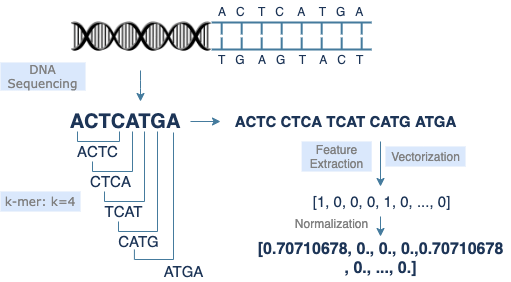 Abbildung 1.
Abbildung 1.
k-mer
Die k-mer-Methode wird häufig in der Vorverarbeitung von DNA-Sequenzen verwendet. Sie extrahiert einen kleinen Abschnitt der Länge k, beginnend bei jeder Base der ursprünglichen Sequenz, und wandelt dadurch eine lange Sequenz der Länge s in (s-k+1) kurze Sequenzen der Länge k um. Die Anpassung des Werts von k verbessert die Modellleistung. Listen kurzer Sequenzen sind einfacher für das Einlesen von Daten, die Merkmalsextraktion und die Vektorisierung.
Vektorisierung
DNA-Sequenzen werden in Textform vektorisiert. Eine durch k-mer transformierte Sequenz wird zu einer Liste kurzer Sequenzen, die wie eine Liste einzelner Wörter in einem Satz aussieht. Daher sollten die meisten Modelle der Verarbeitung natürlicher Sprache auch für DNA-Sequenzdaten funktionieren. Ähnliche Methodologien können auf Modelltraining, Merkmalsextraktion und Kodierung angewendet werden. Da jedes Modell seine eigenen Vorteile und Nachteile hat, hängt die Auswahl der Modelle von den Merkmalen der Daten und dem Forschungszweck ab. Zum Beispiel implementiert CountVectorizer, ein Bag-of-Words-Modell, die Merkmalsextraktion durch unkomplizierte Tokenisierung. Es setzt keine Begrenzung für die Datenlänge, aber das zurückgegebene Ergebnis ist im Hinblick auf den Ähnlichkeitsvergleich weniger offensichtlich.
Milvus-Demo
Milvus kann unstrukturierte Daten problemlos verwalten und unter Billionen von Vektoren innerhalb einer durchschnittlichen Verzögerung von Millisekunden die ähnlichsten Ergebnisse abrufen. Seine Ähnlichkeitssuche basiert auf dem Suchalgorithmus Approximate Nearest Neighbor (ANN). Diese Highlights machen Milvus zu einer großartigen Option, um Vektoren von DNA-Sequenzen zu verwalten, und fördern somit die Entwicklung und Anwendungen der Bioinformatik.
Hier ist eine Demo, die zeigt, wie man ein System zur Klassifizierung von DNA-Sequenzen mit Milvus erstellt. Der experimentelle Datensatz umfasst 3 Organismen und 7 Genfamilien. Alle Daten werden mithilfe von k-mers in Listen kurzer Sequenzen umgewandelt. Mit einem vortrainierten CountVectorizer-Modell kodiert das System anschließend Sequenzdaten in Vektoren. Das folgende Flussdiagramm zeigt die Systemstruktur sowie die Prozesse des Einfügens und Suchens.
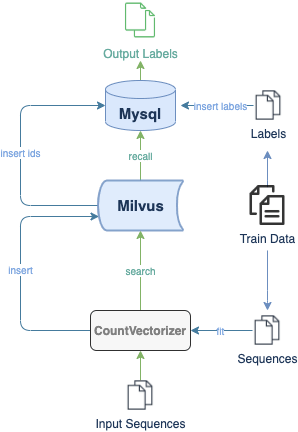 Abbildung 2.
Abbildung 2.
Probieren Sie diese Demo im Milvus bootcamp aus.
In Milvus erstellt das System eine Collection und fügt entsprechende Vektoren von DNA-Sequenzen in die Collection ein (oder in eine Partition, falls aktiviert). Beim Empfang einer Abfrageanforderung gibt Milvus die Distanzen zwischen dem Vektor der eingegebenen DNA-Sequenz und den ähnlichsten Ergebnissen in der Datenbank zurück. Die Klasse der Eingabesequenz und die Ähnlichkeit zwischen DNA-Sequenzen können anhand der Vektordistanzen in den Ergebnissen bestimmt werden.
# Insert vectors to Milvus collection (partition "human")
DNA_human = collection.insert([human_ids, human_vectors], partition_name='human')
# Search topK results (in partition "human") for test vectors
res = collection.search(test_vectors, "vector_field", search_params, limit=topK, partition_names=['human'])
for results in res:
res_ids = results.ids # primary keys of topK results
res_distances = results.distances # distances between topK results & search input
Klassifizierung von DNA-Sequenzen Die Suche nach den ähnlichsten DNA-Sequenzen in Milvus könnte auf die Genfamilie einer unbekannten Probe schließen lassen und somit Aufschluss über ihre mögliche Funktionalität geben. Wenn eine Sequenz als GPCRs klassifiziert wird, hat sie wahrscheinlich Einfluss auf Körperfunktionen. In dieser Demo hat Milvus das System erfolgreich in die Lage versetzt, die Genfamilien der menschlichen DNA-Sequenzen zu identifizieren, mit denen gesucht wurde.
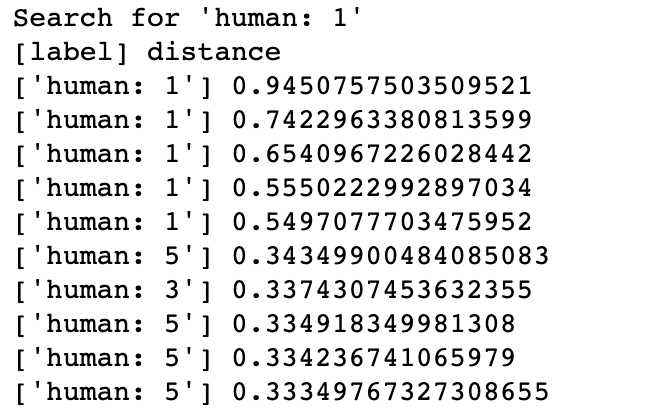 Abbildung 3.
Abbildung 3.
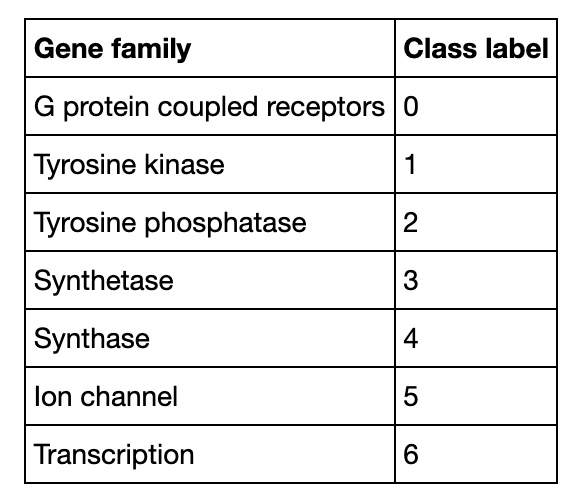 Abbildung 4.
Abbildung 4.
Genetische Ähnlichkeit
Die durchschnittliche Ähnlichkeit von DNA-Sequenzen zwischen Organismen veranschaulicht, wie nahe sich ihre Genome sind. Die Demo sucht in menschlichen Daten jeweils nach DNA-Sequenzen, die denen von Schimpanse und Hund am ähnlichsten sind. Anschließend berechnet und vergleicht sie die durchschnittlichen Inner-Product-Distanzen (0,97 für Schimpanse und 0,70 für Hund), was beweist, dass der Schimpanse ähnlichere Gene mit dem Menschen teilt als der Hund. Mit komplexeren Daten und einem komplexeren Systemdesign ist Milvus in der Lage, genetische Forschung sogar auf einer höheren Ebene zu unterstützen.
search_params = {"metric_type": "IP", "params": {"nprobe": 20}}
Leistung
Die Demo trainiert das Klassifizierungsmodell mit 80 % der menschlichen Probendaten (insgesamt 3629) und verwendet den Rest als Testdaten. Sie vergleicht die Leistung des DNA-Sequenzklassifizierungsmodells, das Milvus verwendet, mit einem Modell, das von Mysql und 5 beliebten Machine-Learning-Klassifikatoren unterstützt wird. Das auf Milvus basierende Modell übertrifft seine Gegenstücke in der Genauigkeit.
from sklearn.model_selection import train_test_split
X, y = human_sequence_kmers, human_labels
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
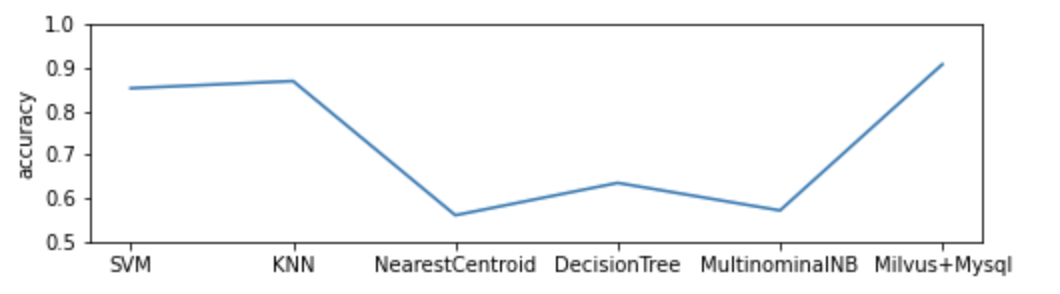 Abbildung 5.
Abbildung 5.
Weitere Erkundung
Mit der Entwicklung der Big-Data-Technologie wird die Vektorisierung von DNA-Sequenzen eine wichtigere Rolle in der genetischen Forschung und Praxis spielen. In Kombination mit Fachwissen in der Bioinformatik können entsprechende Studien zusätzlich von der Einbindung der DNA-Sequenzvektorisierung profitieren. Daher kann Milvus in der Praxis bessere Ergebnisse liefern. Je nach unterschiedlichen Szenarien und Nutzeranforderungen zeigen die von Milvus unterstützte Ähnlichkeitssuche und Distanzberechnung großes Potenzial und viele Möglichkeiten.
- Unbekannte Sequenzen untersuchen: Einigen Forschern zufolge kann die Vektorisierung DNA-Sequenzdaten komprimieren. Gleichzeitig erfordert es weniger Aufwand, Struktur, Funktion und Evolution unbekannter DNA-Sequenzen zu untersuchen. Milvus kann eine enorme Anzahl von DNA-Sequenzvektoren speichern und abrufen, ohne an Genauigkeit zu verlieren.
- Geräte anpassen: Durch traditionelle Algorithmen des Sequenzalignments eingeschränkt, kann die Ähnlichkeitssuche kaum von Verbesserungen der Geräte (CPU/GPU) profitieren. Milvus, das sowohl reguläre CPU-Berechnung als auch GPU-Beschleunigung unterstützt, löst dieses Problem mit einem Algorithmus für approximative nächste Nachbarn.
- Viren erkennen & Ursprünge nachverfolgen: Wissenschaftler haben Genomsequenzen verglichen und berichtet, dass das COVID19-Virus wahrscheinlich von Fledermäusen stammt und zu SARS-COV gehört. Auf Grundlage dieser Schlussfolgerung können Forscher die Stichprobengröße erweitern, um mehr Belege und Muster zu gewinnen.
- Krankheiten diagnostizieren: Klinisch könnten Ärzte DNA-Sequenzen von Patienten und einer gesunden Gruppe vergleichen, um variante Gene zu identifizieren, die Krankheiten verursachen. Es ist möglich, Merkmale zu extrahieren und diese Daten mithilfe geeigneter Algorithmen zu codieren. Milvus ist in der Lage, Distanzen zwischen Vektoren zurückzugeben, die mit Krankheitsdaten in Beziehung gesetzt werden können. Neben der Unterstützung der Krankheitsdiagnose kann diese Anwendung auch dazu beitragen, die Erforschung der zielgerichteten Therapie anzuregen.
Mehr über Milvus erfahren
Milvus ist ein leistungsstarkes Tool, das eine breite Palette von Anwendungen für künstliche Intelligenz und Vektor-Ähnlichkeitssuche unterstützen kann. Um mehr über das Projekt zu erfahren, sehen Sie sich die folgenden Ressourcen an:
Weiterlesen

A Developer's Guide to Exploring Milvus 2.6 Features on Zilliz Cloud
Milvus 2.6 marks a shift from “vector search + glue code” to a more advanced retrieval engine, and it is now Generally Available (GA) on Zilliz Cloud (a managed Milvus service).

Milvus 2.6.x Now Generally Available on Zilliz Cloud, Making Vector Search Faster, Smarter, and More Cost-Efficient for Production AI
Milvus 2.6.x is now GA on Zilliz Cloud, delivering faster vector search, smarter hybrid queries, and lower costs for production RAG and AI applications.

Zilliz Cloud Now Available in Azure North Europe: Bringing AI-Powered Vector Search Closer to European Customers
The addition of the Azure North Europe (Ireland) region further expands our global footprint to better serve our European customers.